Hoy, como una nueva patología extraña, me gustaria presentar este síndrome que buscando en internet, de repente me aparece en la revista Muy Interesante. El síndrome de Tourette, es un trastorno neuropsiquiátrico grave y crónico, que se compone por una serie de tics que la persona que lo padece repite de forma descontrolada. Estos tics tienen la peculiar característica de que en la mayor parte de los casos, son terriblemente obscenos y pueden provocar en la persona enferme una importante inadaptabilidad social.
Los tics de la enfermedad de Tourette pueden ser desde simples contracciones musculares aisladas, resoplidos, o parpadeos, hasta pataletas, insultos, e incluso agresiones. Se supone que este trastorno hereditario puede estar causado por alteraciones neuroendócrinas que involucren a hormonas como la serotonina, la dopamina o la norepinefrina, pero su verdadera etiología todavia sigue en entredicho.
El tratamiento mas habitual, por otra parte, suele ser psicológico, puesto que se ha demostrado ser uno de los más efectivos contra las muchas manifestaciones clínicas añadidas que este síndrome presenta (ansiedad, estrés, déficit de atención...), no obstante en ocasiones se puede llegar a recurrir a tratamiento farmacológico basado en medicación neuroléptica.
Referencias:
Natalia Bermejo Rubio. El síndrome de tourette. [Internet: www.webconsultas.com ] Consultado el 30/11/2014. LINK: http://www.webconsultas.com/sindrome-de-tourette/sindrome-de-tourette-2458
domingo, 30 de noviembre de 2014
Proyecto Enki
Hoy día 30 de noviembre a las 11 de la mañana en la Ciudad deportiva de Riazor (A Coruña), se llevo a cabo la primera carrera de obstáculos por la integración, esta es una iniciativa benéfica llevada a cabo por la FEGEREC (Federación Galega de Enfermidades Raras e Crónicas) cuyo lema no deja indiferente a nadie "solo lo raro es conocido".
La carrera no fue competitiva, en ella pudieron participar todos los públicos, niños, mayores, familias, deportistas, en fin toda persona con o sin discapacidad, que quisiera disfrutar y participar en esta iniciativa, en la que seguro que primó la diversión y el trabajo en equipo. El 100% de los fondos recaudados fueron destinados a favor de los colectivos que luchan por y para la defensa de las personas con diversidad funcional y su integración a través de la práctica de actividades deportivas.
Todo el recorrido estuvo adaptado y preparado para que todo el mundo (discapacitado o no) pudiera superarlo, la carrera consistió en pruebas por equipos de dos integrantes, que superaron con éxito la carrera. Os adjunto un vídeo donde podréis comprobar que un acto de solidaridad no tiene por que no ir ligado a la diversión.
Para mas información: FEGEREC
La carrera no fue competitiva, en ella pudieron participar todos los públicos, niños, mayores, familias, deportistas, en fin toda persona con o sin discapacidad, que quisiera disfrutar y participar en esta iniciativa, en la que seguro que primó la diversión y el trabajo en equipo. El 100% de los fondos recaudados fueron destinados a favor de los colectivos que luchan por y para la defensa de las personas con diversidad funcional y su integración a través de la práctica de actividades deportivas.
Todo el recorrido estuvo adaptado y preparado para que todo el mundo (discapacitado o no) pudiera superarlo, la carrera consistió en pruebas por equipos de dos integrantes, que superaron con éxito la carrera. Os adjunto un vídeo donde podréis comprobar que un acto de solidaridad no tiene por que no ir ligado a la diversión.
Para mas información: FEGEREC
Embarazo Psicológico o pseudociesis
Se trata de un trastorno que afecta a mujeres de todas las edades y que cursa con los síntomas propios de un embarazo natural, como la amenorrea (falta de menstruación), náuseas y vómitos, producción de leche, sensación de movimiento del feto... en definitiva, el cuerpo simula que está embarazado como respuesta a un estímulo emocional. Este estímulo puede darse por el deseo de estar embarazada, o, incluso, por no poder tener hijos y el estrés que eso puede llegar a conllevar.
 Este tipo de embarazos no suponen ningún problema de cara a embarazos reales, lo importante en estos casos es la detección del trastorno, simplemente comprobando el tamaño y situación del útero, y convencer a la mujer de su estado, cuando eso pase el cuerpo recobrará el equilibrio y volverá a la normalidad.
Este tipo de embarazos no suponen ningún problema de cara a embarazos reales, lo importante en estos casos es la detección del trastorno, simplemente comprobando el tamaño y situación del útero, y convencer a la mujer de su estado, cuando eso pase el cuerpo recobrará el equilibrio y volverá a la normalidad.
Como dije antes se trata de un trastorno de tipo somatomorfo, la persona reprime sus sentimientos, puede ser por miedo a defraudar a su familia al no poder quedarse embarazada, o, muchas veces en gente joven, el temor a tenerlos, y estos se canalizan por medio de su cuerpo.
Este trastorno también puede llegar a presentarse en los hombres, cuando sus parejas estan realmente embarazadas, en estos casos la mejor solución es la ayuda psicológica. En el caso masculino recibe el nombre de Síndrome de Couvade, y es un 15% más probable en casos en los que su pareja tiene un embarazo de riesgo.
Una vez solucionado el problema, no debemos pensar que ya ha acabado, puesto que esto indica un cierto desorden psicológico que ha de ser trabajado por el paciente.
sábado, 29 de noviembre de 2014
Enfermedad de las manos frías
La entrada de hoy esta inspirada por una compañera del hospital que la padece, al llegar a casa me he puesto a investigar y aquí esta el resultado, también se denomina fenómeno de Raynaud.
Es un trastorno que se desencadena principalmente por el frío o el estrés, afecta a los capilares de las manos y pies, estos capilares se estrechan, por lo que la sangre no puede llegar a la superficie de la piel, una vez que se restablece el flujo sanguíneo, la piel se enrojece y da una sensación de hormigueo o palpitación. En casos muy graves puede llegar a causar llagas, úlceras, necrosis de los tejidos o gangrena, y como consecuencia de ello la amputación de los dedos.
 Las causas de este fenómeno son desconocidas,
Las causas de este fenómeno son desconocidas,
aunque existe una teoría que lo relaciona con un aumento de los glóbulos rojos. Lo que si se sabe es que hay una serie de factores de riesgo hacia la aparición de este fenómeno, en hombres influye el consumo de tabaco y en mujeres el consumo de alcohol. Para ambos sexos influye la aparición de una bacteria estomacal denominada Helicobacter pylori.
No existe un diagnostico y tratamiento específicos, solo que en los meses de invierno es recomendable el uso de guantes y si el médico lo considera necesario un tratamiento con antihipertesivos para facilitar la reducción de la constricción en los vasos sanguíneos.
Para mas información: http://www.taringa.net/posts/salud-bienestar/3190400/Enfermedad-de-Raynaud-Manos-Frias.html
Es un trastorno que se desencadena principalmente por el frío o el estrés, afecta a los capilares de las manos y pies, estos capilares se estrechan, por lo que la sangre no puede llegar a la superficie de la piel, una vez que se restablece el flujo sanguíneo, la piel se enrojece y da una sensación de hormigueo o palpitación. En casos muy graves puede llegar a causar llagas, úlceras, necrosis de los tejidos o gangrena, y como consecuencia de ello la amputación de los dedos.
Las causas de este fenómeno son desconocidas,aunque existe una teoría que lo relaciona con un aumento de los glóbulos rojos. Lo que si se sabe es que hay una serie de factores de riesgo hacia la aparición de este fenómeno, en hombres influye el consumo de tabaco y en mujeres el consumo de alcohol. Para ambos sexos influye la aparición de una bacteria estomacal denominada Helicobacter pylori.
No existe un diagnostico y tratamiento específicos, solo que en los meses de invierno es recomendable el uso de guantes y si el médico lo considera necesario un tratamiento con antihipertesivos para facilitar la reducción de la constricción en los vasos sanguíneos.
Para mas información: http://www.taringa.net/posts/salud-bienestar/3190400/Enfermedad-de-Raynaud-Manos-Frias.html
Síndrome de Wolfram.
El síndrome de Wolfram es una extraña patología extremadamente rara (con incidencia de 0.0000013) sin cura, y de herencia autosómica recesiva. Engloba dos problemas principalmente: por una parte la Diabetes Mellitus, que aparece durante los primeros diez años de vida, y por otra la atrofia ocular degenerativa. Además, suele también acompañarse de una serie de complicaciones habituales como depresión, psicosis, agresividad, sordera neurosensorial, diabetes insípida o muerte prematura (ya que la mayoría de personas que padecen esta enfermedad no suelen superar los 35 años).
Cito vídeo de una entrevista a un joven de 29 años que padece un curioso caso de S. de Wolfram compuesto por 8 diagnósticos diferentes.
Referencia:
Asociación Española para la Investigación y ayuda del Síndrome de Wolfram. 2014 [Internet]. Consultado el 29/11/2014. LINK: http://aswolfram.org/
Cito vídeo de una entrevista a un joven de 29 años que padece un curioso caso de S. de Wolfram compuesto por 8 diagnósticos diferentes.
Referencia:
Asociación Española para la Investigación y ayuda del Síndrome de Wolfram. 2014 [Internet]. Consultado el 29/11/2014. LINK: http://aswolfram.org/
Dispraxia
En la entrada de hoy voy a hablaros sobre la dispraxia, o trastorno del desarrollo de la coordinación motora (TDAC), enfermedad poco conocida que te puede llevar a pensar que tu amigo simplemente es un poco lentito.
Se trata de una alteración psicomotriz que hace que acciones tan cotidianas como atarse los zapatos o montar en bicicleta se conviertan en gestos lentos y de considerable dificultad.
Esta enfermedad afecta a entre un 2 y 5% de los niños, empieza a manifestarse en el primer año de edad, aciéndose más evidente entre los 5 y 11 años.
La dispraxia afecta prinicipalmente a la coordinación motora, pero también puede afectar a diferentes niveles, créando diferentes tipos de dispraxia:
Se trata de una alteración psicomotriz que hace que acciones tan cotidianas como atarse los zapatos o montar en bicicleta se conviertan en gestos lentos y de considerable dificultad.
Esta enfermedad afecta a entre un 2 y 5% de los niños, empieza a manifestarse en el primer año de edad, aciéndose más evidente entre los 5 y 11 años.
La dispraxia afecta prinicipalmente a la coordinación motora, pero también puede afectar a diferentes niveles, créando diferentes tipos de dispraxia:
- Dispraxia ideomotora: implica dificultad a la hora de crear una secuencia pensamiento - acción en actos sencillos. Interviene en acciones como coger un objeto.
- Dispraxia ideatoria: es similar a la anterior pero afecta a la realización de actos más complejos, como pueden ser atarse los zapatos o coser un botón.
- Dispraxia oromotora o del habla: afecta a los músculos que se encargan de la fonación, teniendo problemas para pronunciar palabras.
- Dispraxia constructiva: dificulta la compresión de los espacios, el individuo afectado tendrá problemas a la hora de comprender relaciones como meter una caja grande en otra más pequeña.
 |
| Daniel Radcliffe caracterizado como Harry Potter. |
Los individuos que presentan este trastorno pueden llevar una vida completamente normal o, incluso, sobresaliente, como le pasa a Daniel Radcliffe, actor que dió vida a Harry Potter, de niño se le detectó la llamada "enfermedad del niño torpe", más concretamente dispraxia ideatoria, lo que no impidió que alcanzara la fama como actor, aunque, aún hoy en día, no es capaz de acciones como atarse los zapatos.
Para tratar esta enfermedad se utilizará un enfoque multidisciplinar, necesitando fisioterapeutas, logopedas o neuropsicólogos. Será muy importante el uso de técnicas de refuerzo por parte de los padres del niño, como puede ser tocar algún instrumento para las dispraxias motoras o movimientos mandibulares para las oromotoras, además de fomentar su autoestima, elemental para que el niño vea que este trastorno no supone ningùn tipo de problema.
viernes, 28 de noviembre de 2014
Hipersexualidad
 Puede que si el post de hoy lo hubiese titulado ninfomanía a todos os hubiera sonado más, pero este término no es del todo correcto, ya que la causa del aumento repentino de la líbido o del deseo sexual no es solo una simple adicción, de hecho, no extiste un consenso en la causa de este trastorno y hay muchas circunstancias que pueden llevar a esta situación, como el consumo de drogas o ciertos medicamentos. Se trata de una enfermedad poco habitual, se estima que solo el 6 % de la población lo sufre y de ese porcentaje únicamente el 2% son mujeres, en contra de lo que se suele pensar.
Puede que si el post de hoy lo hubiese titulado ninfomanía a todos os hubiera sonado más, pero este término no es del todo correcto, ya que la causa del aumento repentino de la líbido o del deseo sexual no es solo una simple adicción, de hecho, no extiste un consenso en la causa de este trastorno y hay muchas circunstancias que pueden llevar a esta situación, como el consumo de drogas o ciertos medicamentos. Se trata de una enfermedad poco habitual, se estima que solo el 6 % de la población lo sufre y de ese porcentaje únicamente el 2% son mujeres, en contra de lo que se suele pensar.Los individuos con este trastorno en muchos casos han sufrido una represión marcada de su sexualidad durante la infancia. La vida de un afectado por la hipersexualidad gira en torno al sexo y a todos los medios que le lleven a él, lo que le produce muchos problemas económicos, sociales y familiares, además de conllevar, en muchos casos, sentimientos de culpa y vergüenza.
Antigüamente existían dos términos diferentes según el sexo del enfermo, ninfomanía, si se trataba de una mujer, y satiriasis, si hablabamos de un hombre, definiéndose como un deseo sexual irrefenable, denominación para la cual, incluso hoy en día, es muy difícil trazar una línea que separe lo normal de lo patológico, habiendo autores que niegan la existencia de este trastorno. Para los que sí reconocen en estos individuos una patología trazan esa línea en el momento en el cual este deseo, este aumento de la líbido, causa incomodidad o impide el funcionamiento social.
El tratamiento de esta enfermedad es, basicamente, psicológico, ayudando al individuo a sobrellevar este trastorno canalizando la energía sexual mediante otras actividades que lo evadan.
En conclusión, la hipersexualidad es un trastorno complicado, un tanto subjetivo y abierto a debate. Lo que sí está claro es que la persona que la sufre no es un obseso, ni un maníaco, simplemente tiene una enfermedad que necesita ser comprendida.
Síndrome de Adie.
El síndrome de Adie (William John Adie) o síndrome de Holmes-Adie, es un trastorno que afecta al sistema nervioso autónomo de inervación ocular y a la pupila. Esta curiosa neuropatía tiene la peculiar caracteristica de que provoca una midriasis (dilatación) de la pupila, lo que se conoce como "pupila tónica de Adie", y muy a menudo viene acompañada de una pérdida de los reflejos tendinosos profundos y una diaforesis aumentada. Su etiología se supone que tiene una base infecciosa, bacterias o virus pueden provocar lesiones en las neuronas postganglionares del SNA parasímpatico del ojo que desencadenen la patología.
 La pupila tónica suele provocar molestias a la hora de fijar la visión en algo o al exponerse a cambios repentinos de luminosidad, por eso se suele recomendar el uso de gafas de lectura / gafas de sol junto con tratamiento farmacológico con Pilocarpina. En el caso de la diaforesis y la pérdida de reflejos la solución se complica: en la primera, sería necesario recurir a la simpatectomia torácica (si ante al tratamiento farmacológico no se responde) y en la segunda, desafortunadamente no tendríamos por el momento ninguna solución.
La pupila tónica suele provocar molestias a la hora de fijar la visión en algo o al exponerse a cambios repentinos de luminosidad, por eso se suele recomendar el uso de gafas de lectura / gafas de sol junto con tratamiento farmacológico con Pilocarpina. En el caso de la diaforesis y la pérdida de reflejos la solución se complica: en la primera, sería necesario recurir a la simpatectomia torácica (si ante al tratamiento farmacológico no se responde) y en la segunda, desafortunadamente no tendríamos por el momento ninguna solución.
Referencias:
Aliaga Gómez J, Marcos Martin C, Serrano Molina C et. al. (2003). Neuroftalmología. En oftalmología en atención primaria (pag 341-356). Jaén: Formación Alcalá.
La pupila tónica suele provocar molestias a la hora de fijar la visión en algo o al exponerse a cambios repentinos de luminosidad, por eso se suele recomendar el uso de gafas de lectura / gafas de sol junto con tratamiento farmacológico con Pilocarpina. En el caso de la diaforesis y la pérdida de reflejos la solución se complica: en la primera, sería necesario recurir a la simpatectomia torácica (si ante al tratamiento farmacológico no se responde) y en la segunda, desafortunadamente no tendríamos por el momento ninguna solución.Referencias:
Aliaga Gómez J, Marcos Martin C, Serrano Molina C et. al. (2003). Neuroftalmología. En oftalmología en atención primaria (pag 341-356). Jaén: Formación Alcalá.
Anomalía de Ebstein
La anomalía de Ebstein consiste en un defecto en la válvula tricúspide, la cual presenta una ubicación y forma anormales. Esta anomalía es congénita, es decir que aparece al nacer, ya que se presenta a medida que el bebe se desarrolla en el útero materno.
La ubicación anormal de esta válvula provoca que funcione de manera deficiente y que la sangre no vaya por el recorrido correcto. En muchos casos los pacientes con esta anomalía también pueden presentar un estrechamiento de la válvula pulmonar, y un agujero en la pared que separa las aurículas.
Se desconoce cual es la causa de esta enfermedad, pero se cree que esta relacionada con la ingesta de ciertos medicamentos durante el embarazo. Por esta razón no existe una manera de prevenirla, solo advertir al médico durante el embarazo del tratamiento que se esta siguiendo.
Los síntomas pueden aparecer en cualquier momento, cuanto antes aparezcan, mas grave es la enfermedad. Estos síntomas van de leves a muy intensos, abarcan labios y uñas morados (por los bajos niveles de oxígeno en sangre). Si son muy intensos pueden presentar dificultad para respirar. En los niños mayores, los síntomas son tos, fatiga, dificultad para respirar e insuficiencia para crecer.
El diagnóstico de esta enfermedad es una auscultación por parte del médico, el cual escuchara ruidos cardíacos anormales, esto suelo ir acompañado de una radiografía de tórax, una resonancia magnética y una ecografía de corazón. En cuanto al tratamiento depende de la gravedad de la anomalía, pero son la aplicación de oxígeno y medicamentos para ayudar con la insuficiencia cardíaca.

Para más información: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/007321.htm
La ubicación anormal de esta válvula provoca que funcione de manera deficiente y que la sangre no vaya por el recorrido correcto. En muchos casos los pacientes con esta anomalía también pueden presentar un estrechamiento de la válvula pulmonar, y un agujero en la pared que separa las aurículas.
Se desconoce cual es la causa de esta enfermedad, pero se cree que esta relacionada con la ingesta de ciertos medicamentos durante el embarazo. Por esta razón no existe una manera de prevenirla, solo advertir al médico durante el embarazo del tratamiento que se esta siguiendo.
Los síntomas pueden aparecer en cualquier momento, cuanto antes aparezcan, mas grave es la enfermedad. Estos síntomas van de leves a muy intensos, abarcan labios y uñas morados (por los bajos niveles de oxígeno en sangre). Si son muy intensos pueden presentar dificultad para respirar. En los niños mayores, los síntomas son tos, fatiga, dificultad para respirar e insuficiencia para crecer.
El diagnóstico de esta enfermedad es una auscultación por parte del médico, el cual escuchara ruidos cardíacos anormales, esto suelo ir acompañado de una radiografía de tórax, una resonancia magnética y una ecografía de corazón. En cuanto al tratamiento depende de la gravedad de la anomalía, pero son la aplicación de oxígeno y medicamentos para ayudar con la insuficiencia cardíaca.
Para más información: http://www.nlm.nih.gov/medlineplus/spanish/ency/article/007321.htm
jueves, 27 de noviembre de 2014
Síndrome de Zellweger
El síndrome cerebro hepato renal, nombre con el que también se le conoce, tiene una incidencia en la población de 1/25000 y 1/100000 nacidos vivos.
.jpg)
Pertenece al grupo de enfermedades peroxisomales, ya que existe una baja producción o ausencia de los peroxisomas, especialmente en tejidos del hígado y los riñones, estos peroxisomas son los que crean las malformaciones en el cerebro y músculo. Se caracteriza por asociar alteraciones neurológicas graves con malformaciones craneofaciales.
Para el diagnóstico prenatal de este síndrome se realiza un cultivo de ammniocitos para detectar una acumulación de ácidos grasos de cadena larga, esta muestra se centrifugan y si no precipita, la prueba será positiva. Actualmente no existe tratamiento, pero se cree que con suministro de manera temprana de ácidos grasos de omega 3, puede frenar el retraso psicomotor y el deterioro auditivo y visual. No obstante el 70% de los afectados mueren en los tres primeros meses de vida, aunque hay varios casos que han llegado al año de edad y solo casos puntuales llegan a la veintena.
 Los síntomas que presentan los pacientes que la padecen son muy numerosos, los mas importantes son, hipotonía severa, braquicefalia, un aumento del tamaño del hígado, coloración amarillenta de la piel y quistes renales. También pueden tener un retraso del crecimiento dentro del útero materno y una vez que hayan nacido, macrofelia, fontanelas muy abiertas, párpados edematosos y numerosas anomalías faciales. Varias disfunciones orgánicas en pulmones, arco aórtico, anomalías en manos, pies (pie plano) y contracturas en rodillas y caderas entre otras.
Los síntomas que presentan los pacientes que la padecen son muy numerosos, los mas importantes son, hipotonía severa, braquicefalia, un aumento del tamaño del hígado, coloración amarillenta de la piel y quistes renales. También pueden tener un retraso del crecimiento dentro del útero materno y una vez que hayan nacido, macrofelia, fontanelas muy abiertas, párpados edematosos y numerosas anomalías faciales. Varias disfunciones orgánicas en pulmones, arco aórtico, anomalías en manos, pies (pie plano) y contracturas en rodillas y caderas entre otras.
Para mas información: http://www.enfermedades-raras.org/index.php/las-enfermedades-raras/listado-de-patologias?id=1020
Pertenece al grupo de enfermedades peroxisomales, ya que existe una baja producción o ausencia de los peroxisomas, especialmente en tejidos del hígado y los riñones, estos peroxisomas son los que crean las malformaciones en el cerebro y músculo. Se caracteriza por asociar alteraciones neurológicas graves con malformaciones craneofaciales.
Para el diagnóstico prenatal de este síndrome se realiza un cultivo de ammniocitos para detectar una acumulación de ácidos grasos de cadena larga, esta muestra se centrifugan y si no precipita, la prueba será positiva. Actualmente no existe tratamiento, pero se cree que con suministro de manera temprana de ácidos grasos de omega 3, puede frenar el retraso psicomotor y el deterioro auditivo y visual. No obstante el 70% de los afectados mueren en los tres primeros meses de vida, aunque hay varios casos que han llegado al año de edad y solo casos puntuales llegan a la veintena.
Los síntomas que presentan los pacientes que la padecen son muy numerosos, los mas importantes son, hipotonía severa, braquicefalia, un aumento del tamaño del hígado, coloración amarillenta de la piel y quistes renales. También pueden tener un retraso del crecimiento dentro del útero materno y una vez que hayan nacido, macrofelia, fontanelas muy abiertas, párpados edematosos y numerosas anomalías faciales. Varias disfunciones orgánicas en pulmones, arco aórtico, anomalías en manos, pies (pie plano) y contracturas en rodillas y caderas entre otras.Para mas información: http://www.enfermedades-raras.org/index.php/las-enfermedades-raras/listado-de-patologias?id=1020
Síndrome del QT largo.
 El síndrome del QT largo es una curiosa patología que afecta al sistema de conducción electrica del corazón. Esta enfermedad, concretamente involucra a los canales iónicos responsables de la repolarización del miocardio (onda T en el trazado electrocardiográfico), de este modo, incrementa el tiempo que necesita el corazón para volver a preparar sus células para el siguiente latido.A menudo esta patología provoca arritmias caracteristicas conocidas como "torsades de pointes" que pueden llegar incluso a producir pérdidas repentinas en el estado de alerta o incluso síncopes (desmayos).
El síndrome del QT largo es una curiosa patología que afecta al sistema de conducción electrica del corazón. Esta enfermedad, concretamente involucra a los canales iónicos responsables de la repolarización del miocardio (onda T en el trazado electrocardiográfico), de este modo, incrementa el tiempo que necesita el corazón para volver a preparar sus células para el siguiente latido.A menudo esta patología provoca arritmias caracteristicas conocidas como "torsades de pointes" que pueden llegar incluso a producir pérdidas repentinas en el estado de alerta o incluso síncopes (desmayos).Habitualmente estas arritmias son desencadenadas por estímulos fuertes como un susto, o un ruido muy alto y en algunos casos, puede darse la situación de que si la arritmia no se revierte, el paciente entre en fibrilación ventricular.
Referencia:
Fundación SADS. Una guia para pacientes y proveedores de cuidados médicos. [Internet]. Consultado el 27/11/2014. Actualizado el 6/2008. LINK: http://www.sads.org/SADS/media/pdf/SADS-LQTS-Broch-Spanish-09.pdf
Terrores nocturnos
En el post de hoy voy a hablaros de los terrores nocturnos, una enfermedad poco conocida y poco frecuente, solo afecta de un 3 a un 6 % de la población, que puede llevar a que crean que, simplemente, vives demasiado tus pesadillas, y que no se arregla con dejar de ver pelis de miedo a solas.

Se trata de un trastorno del sueño que se dá, sobretodo, en niños de entre 4 y 12 años, y que , aunque no debe ser motivo de preocupación ni denota ninguna enfermedad subyacente, es muy molesto, ya que se sufre verdadero terror. Los terrores nocturnos ocurren durante la fase de sueño profundo, con lo que no se trata realmente de una pesadilla, estas se crean en la fase de sueño no profundo, si no que es una reacción negativa súbita en el cambio de una fase del sueño a otra, entre 2 y 3 horas una vez empezado el sueño.
El individuo, niño en la mayoría de los casos, puede llegar a incorporarse de la cama y moverse agitadamente, además sufrirá un aumento de la frecuencia cardíaca y respiratoria. Pasados unos minutos seguirá durmiendo placidamente y, al día siguiente, no recordarán nada.
La causa de los terrores nocturnos suele ser una hiperactividad del sistema nervioso central (SNC) durante el sueño, esta se puede dar debido a que el organismo está en crecimiento o por haber heredado tendencias hiperactivas.
Lo que está claro es que, para un padre, no es fácil saber como reaccionar cuando un hijo se encuentra en un estado como este, siendo normal que sientan una gran impotencia; lo que debemos hacer ante un caso de terror nocturno es asegurarnos de que el niño no se daña en el proceso; no intentar despertarlo, ya que dichos intentos no suelen funcionar y, si lo hacen, solo crean una sensación de desorientación en el niño y dificultad para dormir.
En cuanto al tratamiento, no hay una solución inmediata ni un fármaco milagroso, simplemente irá pasando por sí solo con el crecimiento del niño, aunque establecer rutinas de sueño-vigilia, acostarse temprano o reducir el estrés al que se pueda ver sometido ayudaran mucho en el proceso.
miércoles, 26 de noviembre de 2014
Gigantismo y acromegalia.
En mi último post os hablé de la causa más común de enanismo, hoy os voy a hablar del extremo contrario, el gigantismo.
Esta enfermedad, se dá por un exceso en la producción de hormona de crecimiento (GH) durante la niñez, si se produce cuando el crecimiento óseo ya ha finalizado nos encontramos ante una enfermedad diferente, la acromegalia.
La sobreproducción de hormonas de crecimiento es producida, en la mayor parte de los casos, debido a la existencia de un tumor benigno de hipófisis, lo cual impide su correcto funcionamiento.
Las hormonas del crecimiento impedirán una osificación normal de los huesos y estos seguirán creciendo, dándose el llamado gigantismo. Además impedirán la secreción normal de hormonas por parte de las gónadas, retrasando en muchos casos la pubertad y dando lugar a una disminución notable de la función sexual. Este tipo de gigantismo no es muy común, recibe el nombre de gigantismo eunucoideo.
Esta enfermedad, se dá por un exceso en la producción de hormona de crecimiento (GH) durante la niñez, si se produce cuando el crecimiento óseo ya ha finalizado nos encontramos ante una enfermedad diferente, la acromegalia.
La sobreproducción de hormonas de crecimiento es producida, en la mayor parte de los casos, debido a la existencia de un tumor benigno de hipófisis, lo cual impide su correcto funcionamiento.
Las hormonas del crecimiento impedirán una osificación normal de los huesos y estos seguirán creciendo, dándose el llamado gigantismo. Además impedirán la secreción normal de hormonas por parte de las gónadas, retrasando en muchos casos la pubertad y dando lugar a una disminución notable de la función sexual. Este tipo de gigantismo no es muy común, recibe el nombre de gigantismo eunucoideo.
Síndrome de Tourette
Este síndrome también se conoce con el nombre de enfermedad de Gilles; afecta más a los niños que a las niñas, y los primeros síntomas aparecen entre los 7 y 10 años de edad. Consiste en un trastorno neurológico que empuja a la persona que lo padece a realizar sonidos y movimientos de forma involuntaria, denominados tic´s que pueden ser simples o complejos, y que aumentan en situaciones de estrés. También pueden presentar un trastorno obsesivo-compulsivo o déficit de atención, depresión y ansiedad.
Las causas actualmente son desconocidas, pero se cree que podría estar relacionado con ciertas zonas del cerebro y alteraciones en las sustancias químicas. Lo que sí que se sabe es que se trata de un trastorno hereditario.
En cuanto al diagnóstico es únicamente observacional, ya que las pruebas de laboratorio no lo desvelan; se observa la aparición, duración, complejidad, tipo y severidad de tic´s motores y fónicos que no sean justificados por otro tipo de patología o por fármacos que consuma el paciente. Estos tic´s se tratan con fármacos neurolépticos y con terapia psicológica.
Para mas información: http://www.webconsultas.com/sindrome-de-tourette/sindrome-de-tourette-2458
Las causas actualmente son desconocidas, pero se cree que podría estar relacionado con ciertas zonas del cerebro y alteraciones en las sustancias químicas. Lo que sí que se sabe es que se trata de un trastorno hereditario.
En cuanto al diagnóstico es únicamente observacional, ya que las pruebas de laboratorio no lo desvelan; se observa la aparición, duración, complejidad, tipo y severidad de tic´s motores y fónicos que no sean justificados por otro tipo de patología o por fármacos que consuma el paciente. Estos tic´s se tratan con fármacos neurolépticos y con terapia psicológica.
Para mas información: http://www.webconsultas.com/sindrome-de-tourette/sindrome-de-tourette-2458
La enfermedad de Tay-Sachs.
Esta patología, en muchas ocasiones mortal, es un tipo de enfermedad genética autosómica recesiva asociada al cromosoma 15 que afecta al sistema nervioso. La enfermedad de Tay-Sachs, concretamente impide la síntesis de la Hexosaminidasa A, esta proteína es la encargada de eliminar los gangliósidos GM2 que naturalmente se fabrican en el cerebro. Debido a esto, los gangliósidos se siguen sintetizando de forma descontrolada en estas células nerviosas y se acumulan progresivamente.
Normalmente los niños que genéticamente heredan el doble alelo recesivo empiezan a desarrollar la enfermedad ya en el útero materno y esta progresa con una gran rapidez. Se estima que la vida de estos enfermos no suele superar los 4/5 años de edad.
Entre la sintomatologia mas destacada encontramos los deficits sensoriales (sordera y ceguera), mentales y sociales, la irritabilidad y apatia, las crisis epilépticas, el lento crecimiento, la demencia, y uno de los rasgos más característicos de la enfermedad: la pérdida progresiva de la capacidad motora, que empieza con una rápida disminución del tono muscular y culmina con la parálisis total.
Remito un importante link de la Asociación Acción y Cura para Tay Sachs.
Referencia:
Jonhston MV, Neurodegenerative Disorders of Chilhood. Eds. Nelson Textbook of Pediatrics. Edición 18. Philadelphia, Saunders Elsevier 2007. Capítulo 599.
Normalmente los niños que genéticamente heredan el doble alelo recesivo empiezan a desarrollar la enfermedad ya en el útero materno y esta progresa con una gran rapidez. Se estima que la vida de estos enfermos no suele superar los 4/5 años de edad.
Entre la sintomatologia mas destacada encontramos los deficits sensoriales (sordera y ceguera), mentales y sociales, la irritabilidad y apatia, las crisis epilépticas, el lento crecimiento, la demencia, y uno de los rasgos más característicos de la enfermedad: la pérdida progresiva de la capacidad motora, que empieza con una rápida disminución del tono muscular y culmina con la parálisis total.
Remito un importante link de la Asociación Acción y Cura para Tay Sachs.
Referencia:
Jonhston MV, Neurodegenerative Disorders of Chilhood. Eds. Nelson Textbook of Pediatrics. Edición 18. Philadelphia, Saunders Elsevier 2007. Capítulo 599.
martes, 25 de noviembre de 2014
Síndrome de Marfan
Este síndrome consiste en un trastorno del tejido conectivo (el que fortalece las estructuras corporales) lo que afecta de manera diferente al esqueleto, al sistema cardiovascular, ojos y piel. Es causado por un defecto en un gen llamado fibrilina-1, es un síndrome hereditario, pero en un 30% de los casos no existen antecedentes.

 En el esqueleto causa crecimiento excesivo de los huesos largos, por lo que las personas que lo padecen tienen una gran estatura, al igual que sus manos y piernas largas, pie plano, un torax que se hunde o sobresale, mandíbula pequeña y baja, cara estrecha y delgada y escoliosis.
En el esqueleto causa crecimiento excesivo de los huesos largos, por lo que las personas que lo padecen tienen una gran estatura, al igual que sus manos y piernas largas, pie plano, un torax que se hunde o sobresale, mandíbula pequeña y baja, cara estrecha y delgada y escoliosis.
En el sistema cardiovascular, afectan al tejido pulmonar y puede debilitar la aorta.
También pueden presentar miopía, hipotonía y dificultades en el aprendizaje.
Las pruebas y exámenes que se llevan a cabo son exámenes físicos rutinarios de las articulaciones, así como comprobar si existen signos de aneurisma, problemas con las válvulas cardíacas (por lo que se realiza anualmente un electrocardiograma) y atelectasia pulmonar.
En cuanto al tratamiento consiste en medicamentos para disminuir la frecuencia cardíaca, vigilar la escoliosis sobre todo durante la adolescencia, tratar los problemas visuales cuando sea posible, vigilar y tener en cuenta las posibles complicaciones.
Para mas información: http://www.nlm.nih.g
ov/medlineplus/spanish/ency/article/000418.htm
En el esqueleto causa crecimiento excesivo de los huesos largos, por lo que las personas que lo padecen tienen una gran estatura, al igual que sus manos y piernas largas, pie plano, un torax que se hunde o sobresale, mandíbula pequeña y baja, cara estrecha y delgada y escoliosis.En el sistema cardiovascular, afectan al tejido pulmonar y puede debilitar la aorta.
También pueden presentar miopía, hipotonía y dificultades en el aprendizaje.
Las pruebas y exámenes que se llevan a cabo son exámenes físicos rutinarios de las articulaciones, así como comprobar si existen signos de aneurisma, problemas con las válvulas cardíacas (por lo que se realiza anualmente un electrocardiograma) y atelectasia pulmonar.
En cuanto al tratamiento consiste en medicamentos para disminuir la frecuencia cardíaca, vigilar la escoliosis sobre todo durante la adolescencia, tratar los problemas visuales cuando sea posible, vigilar y tener en cuenta las posibles complicaciones.
Para mas información: http://www.nlm.nih.g
ov/medlineplus/spanish/ency/article/000418.htm
La insensibilidad congénita al dolor.
La insensibilidad congénita al dolor o CIPA (Congenital Insensibility to Pain Anhidrosis) es una curiosa patología hereditaria (autosómica recesiva) que se supone afecta a zonas específicas del sistema nerviosa involucradas en la percepción del dolor y la temperatura. Las personas que padecen esta enfermedad se cree que poseen un defecto genético en las células nerviosas encargadas de transmitir estas señales, debido a esto, a menudo sufren lesiones como traumatismos o quemaduras cuando son más pequeños. Otros expertos sugieren que el trastorno tiene un origen endocrino, es decir, que el cuerpo de los enfermos produce una cantidad exorbitante de endorfinas (analgésicos naturales), lo cual inmuniza a la persona frente a estos desagradables estímulos.
La CIPA es una enfermedad extremadamente rara, tan solo han sido documentados 60 casos en EEUU y entorno a los 300 en Japón. Desgraciadamente, muchos de los enfermos de CIPA perecen a edades tempranas por su incapacidad de producir sudor (anhidrosis) que regule su temperatura corporal.
Bibliografía:
Dr. Byron Orlando Albuja Echeverría. Insensibilidad congénita al dolor con anhidrosis. Diagnóstico clínico, evolucion y complicaciones. Reporte de un caso [Internet]. Consulta el 24/11/2014. Actualizado el 9/4/2014. LINK: http://www.scielo.org.ar/scielo.php?pid=S0325-00752014000500014&script=sci_arttext
Pablo Muriedas. Insensibilidad congénita al dolor. El infierno de no sufrif [Internet]. Consulta el 24/11/2014. Actualizado el 3/1/2014. LINK:http://www.gonzoo.com/zoom/story/insensibilidad-congenita-al-dolor-el-infierno-de-no-sufrir-1249/
Acondroplasia
 |
| El actor Peter Dinklage. Fuente: GQ revista |
La acondroplasia es la causa del 70% de los casos de enanismo, En esta enfermedad se da un crecimiento anormalmente lento del cartílago a partir del cual se creará el hueso. Todos los huesos largos se quedarán cortos, pero el crecimiento en anchura continuara normalmente. Esto dará lugar al llamado "enanismo desproporcionado", con un tamaño casi normal de la cabeza y del tronco pero miembros inferiores y superiores cortos, el tamaño medio de un hombre acondroplásico es de 1,31 y el de una mujer 1,24. Las líneas cartilagenosas del cráneo también se ven afectadas, por lo que encontramos una raíz nasal muy hundida. Aún así, la capacidad intelectual es completamente normal, lo que puede producir trastornos psicológicos al tener plena consciencia de su enfermedad.
La aparición de esta enfermedad es debida a mutaciones en el material genético, esta mutación podemos heredarla de nuestros progenitores, tratándose de un gen autosómico dominante, por lo tanto, si uno de los padres padece acondroplasia el bebé tendrá un 50% de posibilidades de heredar la enfermedad, siendo la probabilidad de heredarla de un 75% si ambos padres sufren acondroplasia.
Sin embargo, la mayoría de casos de esta enfermedad se dan por mutaciones espontáneas, en estos casos padres sin esta enfermedad pueden engendrar un hijo acondroplásico.
Como es lógico, una alteración así del cuerpo acarrea ciertos problemas, entre ellos contamos con problemas respiratorios como eupneas, ausencia de la respiración de manera momentánea, obesidad e infección de oidos, además de dolores de espalda y articulaciones y trastornos columnares como cifosis, lordosis o compresiones medulares.
Existen diferentes pruebas para detectar esta enfermedad, durante el embarazo un indicador es el exceso de líquido amniótico rodeando el feto, lo que se puede comprobar con una ecografía. En el neonato la hidrocefalia, presencia de líquido en el cráneo que
 |
| Hidrocefalia en neonato con Acondroplasia. Fuente: Acondroplasiabg. |
El tratamiento se basará en paliar y minimizar los problemas derivados de este trastorno, ya que no tiene cura. El individuo con acondroplasia tendrá que lidiar con los problemas físicos y psicológicos de la enfermedad durante toda su vida, en caso de que el gen se herede de ambos padres el individuo no sobrevivirá más allá de unos pocos meses por norma general.
lunes, 24 de noviembre de 2014
Síndrome de Apert
El síndrome de Apert también conocido como Acrocefalosindactilia I, afecta a 1 de cada 100000 casos y consiste en un cierre prematuro de las suturas craneales, las cuales suelen cerrarse aproximadamente a los 2 años, ya que la capacidad craneal a esta edad es casi 4 veces mayor a la del nacimiento.
 Este cierre prematuro trae consigo una serie de alteraciones en los bebes que lo padecen, como malformaciones en el cráneo el cual se vuelve "inexpandible", y su no se trata puede producir hipertensión craneal, retraso mental, ceguera y perdida de audición; alteraciones faciales que afectan a frente, nariz y ojos. También pueden aparecer malformaciones en los dedos de las manos y pies, ya que pueden estar unidos entre ellos o la ausencia de alguno.
Este cierre prematuro trae consigo una serie de alteraciones en los bebes que lo padecen, como malformaciones en el cráneo el cual se vuelve "inexpandible", y su no se trata puede producir hipertensión craneal, retraso mental, ceguera y perdida de audición; alteraciones faciales que afectan a frente, nariz y ojos. También pueden aparecer malformaciones en los dedos de las manos y pies, ya que pueden estar unidos entre ellos o la ausencia de alguno.
El agente causante de este síndrome es una mutación en los factores de crecimiento de los fibroblastos, producida en el proceso de formación de los gametos, no obstante no se conoce la causa de esta mutación, solo que una persona afectada tiene un 50% de posibilidades de transmitírselo a su descendencia y que la frecuencia de dicha mutación en padres no afectados aumenta con la edad del padre.
En cuanto al diagnóstico de este síndrome basta con una radiografía de manos y pies, pero se complementa con un estudio genético. El tratamiento consiste en una operación precoz, aunque el pronóstico depende de cada niño.
Para mas información: http://salud.discapnet.es/Castellano/Salud/Discapacidades/Desarrollo%20Motor/Sindrome%20de%20Apert/Paginas/default.aspx
Este cierre prematuro trae consigo una serie de alteraciones en los bebes que lo padecen, como malformaciones en el cráneo el cual se vuelve "inexpandible", y su no se trata puede producir hipertensión craneal, retraso mental, ceguera y perdida de audición; alteraciones faciales que afectan a frente, nariz y ojos. También pueden aparecer malformaciones en los dedos de las manos y pies, ya que pueden estar unidos entre ellos o la ausencia de alguno.El agente causante de este síndrome es una mutación en los factores de crecimiento de los fibroblastos, producida en el proceso de formación de los gametos, no obstante no se conoce la causa de esta mutación, solo que una persona afectada tiene un 50% de posibilidades de transmitírselo a su descendencia y que la frecuencia de dicha mutación en padres no afectados aumenta con la edad del padre.
En cuanto al diagnóstico de este síndrome basta con una radiografía de manos y pies, pero se complementa con un estudio genético. El tratamiento consiste en una operación precoz, aunque el pronóstico depende de cada niño.
Para mas información: http://salud.discapnet.es/Castellano/Salud/Discapacidades/Desarrollo%20Motor/Sindrome%20de%20Apert/Paginas/default.aspx
La progeria de Hutchinson-Gilford.
 Esta enfermedad se presenta como una patología considerablemente rara que afecta a la velocidad del crecimiento. Cuando los niños afectados alcanzan los 50 cm de altura,comienzan una serie de fluctuaciones en la velocidad de desarrollo corporal que al principio los llevan a crecer entorno a 25 cm por año y luego los ralentizan, incluso mas de lo normal, hasta alcanzar los 4 cm por año.
Esta enfermedad se presenta como una patología considerablemente rara que afecta a la velocidad del crecimiento. Cuando los niños afectados alcanzan los 50 cm de altura,comienzan una serie de fluctuaciones en la velocidad de desarrollo corporal que al principio los llevan a crecer entorno a 25 cm por año y luego los ralentizan, incluso mas de lo normal, hasta alcanzar los 4 cm por año.
Posteriormente, cuando alcanzan la pubertad, su tasa de crecimiento vuelve a dispararse pudiendo llegar a alcanzar una velocidad de envejecimiento 7 veces mayor de lo normal.
Aquellos que sufren este sindrome, además tambien presentan una serie de rasgos comunes como la piel extremadamente fina, la cabeza mas grande de lo normal (en comparación con la cara), ojos muy grandes y saltones, dientes amontonados, o gran calibre en las venas de la cabellera.
Gloria Ugidos Rio, Pilar Valentín Vertomeu. MSc in Bioginformatics (UAB). Referencia web: http://bioinformatica.uab.es/biocomputacio/treballs02-03/Ugidos_Valentin/p%C3%A0gines%20treball/s%C3%ADndrome%20de%20hutchinson.htm (consultado el 23/11/2014).
Enfermedad de Legg-Calve-Perthes
La enfermedad de Legg-Calve-Perthes, o enfermedad de Perthes, es una enfermedad poco común perteneciente al área de la traumatología. Se da en varones de entre 4 a 10 años de edad y las causas son desconocidas.
| Radriografía, degeneración ósea de la cabeza femoral derecha. |
Este trastorno consiste en que la cabeza del fémur no recibe el suficiente aporte sanguíneo, lo que resulta en la muerte de la zona, esta se colapsa y quedará prácticamente plana. Puede darse en ambas piernas aunque no es lo habitual. Pero, no todo son malas noticias ya que, el suministro de sangre vuelve pasados unos meses, produciéndose una regeneración ósea que sustituirá al tejido necrosado en 2 o 3 años,mientras tanto, el niño en cuestión tendrá que ayudarse de instrumentos ortopédicos para poder moverse.
Podemos detectar esta enfermedad a partir de una ligera cojera, acortamiento de la pierna o por la perdida de tono muscular en el muslo de la pierna afectada. En cuanto al dolor, no es una enfermedad especialmente dolorosa en sus inicios, por norma general, aunque si produce un notable desgaste en la rodilla que puede acarrear molestias considerables.
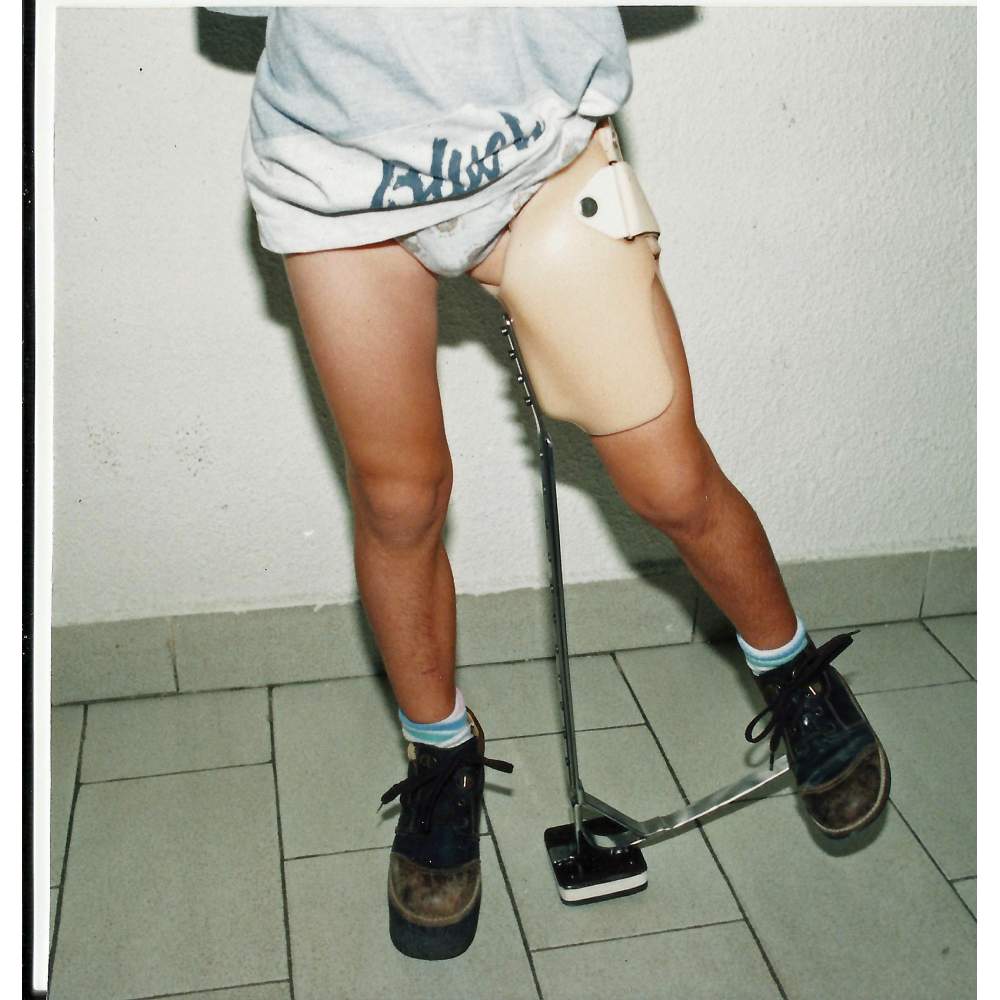 |
| Órtesis específica para enfermedad de Perthes. |
El tratamiento consistirá en la 'contención' del hueso en su cavidad natural, acetábulo, para eso realizaremos un reposo incial para calmar el dolor, continuaremos con una vida relativamente normal pero con restricción de actividades como correr y realizaremos sesiones de fisioterapia para mantener el tono muscular. Esto lo acompañaremos con ayudas ortopédicas, antiinflamatorios y analgesia.
Suscribirse a:
Entradas (Atom)